Esempi di dispositivi medici
Cosa hanno in comune impianti medici, apparecchi per dialisi, pacemaker, prodotti dentali, cerotti e bendaggi, ausili visivi, apparecchiature radiologiche, preservativi, test di gravidanza, strumenti di laboratorio e test COVID-19? Sono tutti dispositivi medici sorvegliati da Swissmedic. Prima di essere immessi in commercio devono soddisfare determinati requisiti e superare una procedura di valutazione della conformità. Sul mercato è possibile trovare fino a mezzo milione di dispositivi diversi.
Esempi di dispositivi medico-diagnostici in vitro
I dispositivi medico-diagnostici in vitro (DIV) sono un sottogruppo dei dispositivi medici. «Un dispositivo medico-diagnostico in vitro è un dispositivo medico per l’esame in vitro di campioni prelevati dal corpo umano, come il sangue o l’urina. Si tratta di test basati ad esempio su analisi biologiche che aiutano a controllare lo stato di salute di una persona: basti pensare ai test per il tumore al seno, per la glicemia o per l’HIV», spiega Evelyn Aeschlimann. «I fabbricanti con sede in Svizzera sono soggetti all’obbligo di notifica quando immettono in commercio per la prima volta un DIV nel territorio elvetico.»
Importanza
Il tema è importante, e lo dimostra il fatto che la Confederazione ha creato una base giuridica specifica e separata per i DIV. «Questa base è costituita da due ordinanze del Consiglio federale con disposizioni legali. Si è trattato di un grande progetto, che ha richiesto mesi di lavoro e l’impegno di numerose persone di diverse autorità», ricorda Andreas Schlegel. La competenza al riguardo spettava all’Ufficio federale della sanità pubblica (UFSP). «Swissmedic si è occupato soprattutto di verificare la fattibilità in fase di esecuzione.»
Accesso al mercato
Sul mercato si contano 500 000 dispositivi medici, di cui circa 40 000 DIV. Per fare un confronto, in Svizzera esistono circa 5700 medicamenti omologati per uso umano. «A differenza dei medicamenti, i dispositivi medici non sono sottoposti a un’omologazione ufficiale, perché risulterebbe eccessivo. Si applica invece il sistema di valutazione della conformità, nell’ambito del quale si dimostra che i requisiti di sicurezza e prestazione vigenti sono soddisfatti e che esiste un rapporto rischi/benefici accettabile», spiega Einat Schmutz. La procedura prevede che per i prodotti ad alto rischio si debba interpellare un organismo designato (OD), la cui designazione e sorveglianza competono a Swissmedic.
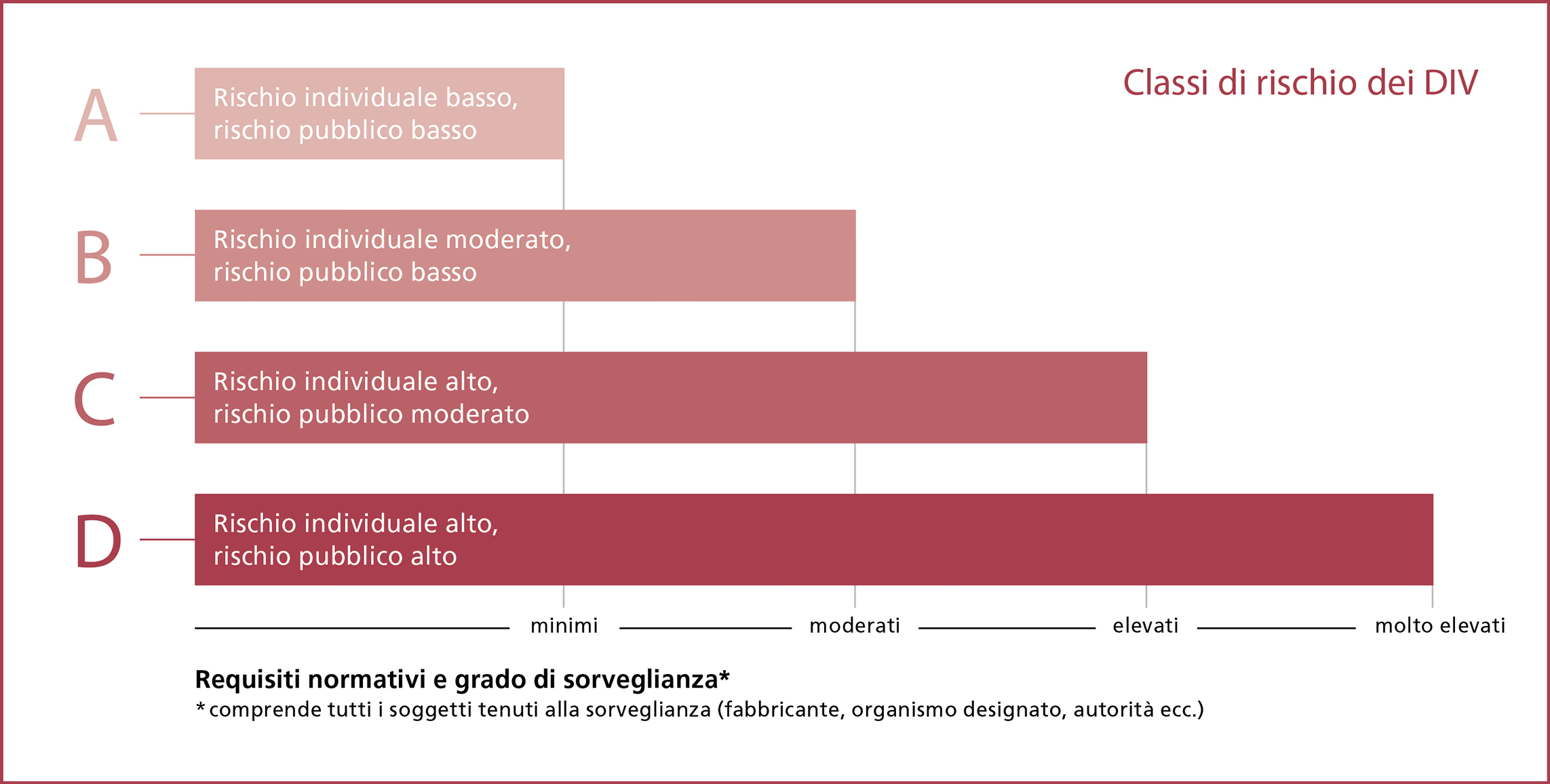
Classi di rischio
Prima dell’immissione in commercio, i DIV vengono assegnati a una classe di rischio specifica (A, B, C, D); la classe A comprende i prodotti a basso rischio (vedi box). «Con questo nuovo sistema a quattro classi, nell’ambito della valutazione della conformità è necessario coinvolgere un organismo designato per un numero molto maggiore di DIV rispetto a prima», spiega Schmutz. Se in passato era sufficiente un’autodichiarazione del fabbricante per immettere sul mercato la maggior parte dei DIV, oggi questo vale solo per i prodotti di classe A, che rappresentano circa il 20 per cento del totale. Per i DIV delle categorie B, C e D occorre una verifica da parte di un organismo designato. L’obiettivo primario è garantire la sicurezza dei prodotti. «Un aspetto fondamentale consiste nella valutazione delle prestazioni cliniche, per la quale è necessaria una raccolta di dati clinici nell’ambito di studi delle prestazioni. La nostra priorità è la sicurezza dei pazienti», spiega Einat Schmutz citando un esempio relativo alla prassi quotidiana: «A causa dei requisiti più severi, i test genetici e tumorali sono passati dalla classe di rischio più bassa alla seconda più alta.»


