Exemples de dispositifs médicaux
Qu’ont en commun les implants médicaux, dialyseurs, pacemakers, produits dentaires, pansements et bandages, lunettes, appareils radiologiques, préservatifs, tests de grossesse, instruments de laboratoire et tests COVID ? Ce sont tous des dispositifs médicaux, qui sont surveillés par Swissmedic. Pour pouvoir être mis sur le marché, ils doivent satisfaire à certaines exigences et passer ce que l’on appelle une procédure d’évaluation de la conformité. Jusqu’à un demi-million de dispositifs peuvent être présents sur le marché.
Exemples de dispositifs de diagnostic in vitro
Les dispositifs de diagnostic in vitro (DIV) sont un sous-groupe de dispositifs médicaux. « Un dispositif de diagnostic in vitro est un dispositif médical destiné à un examen en laboratoire d’échantillons provenant du corps humain, comme le sang ou l’urine. Il s’agit de tests reposant p. ex. sur des examens biologiques et qui aident à surveiller l’état de santé d’une personne, comme les tests de dépistage du cancer du sein ou du VIH, ou encore de glycémie », explique Evelyn Aeschlimann. « Les fabricants ayant leur siège en Suisse doivent déclarer les DIV qu’ils souhaitent commercialiser dans notre pays. »
L’importance des DIV
Les DIV sont suffisamment importants pour que la Confédération ait décidé d’adopter une réglementation qui leur est propre. « La base légale sur laquelle ils reposent est constituée par deux ordonnances du Conseil fédéral. Ça a été un projet de grand ampleur, qui a occupé plusieurs personnes travaillant pour différentes autorités pendant des mois », se souvient Andreas Schlegel. Une procédure dont la direction a été confiée à l’Office fédéral de la santé publique (OFSP). « Swissmedic a été impliqué, en particulier pour contrôler l’applicabilité lors de l’exécution. »
La mise sur le marché
On compte 500 000 dispositifs médicaux – dont environ 40 000 DIV. Par comparaison : quelque 5700 médicaments à usage humain sont autorisés en Suisse. « Contrairement aux médicaments, les dispositifs médicaux ne sont pas soumis à une procédure officielle d’autorisation – ce qui serait démesuré – mais à une évaluation de la conformité. Cette évaluation a pour but de montrer que les exigences de sécurité et de performances applicables sont remplies et que le rapport bénéfice-risque est acceptable », explique Einat Schmutz. Pour les dispositifs présentant un risque élevé, il faut recourir à un organisme désigné, dont la désignation et la surveillance relèvent de la responsabilité de Swissmedic.
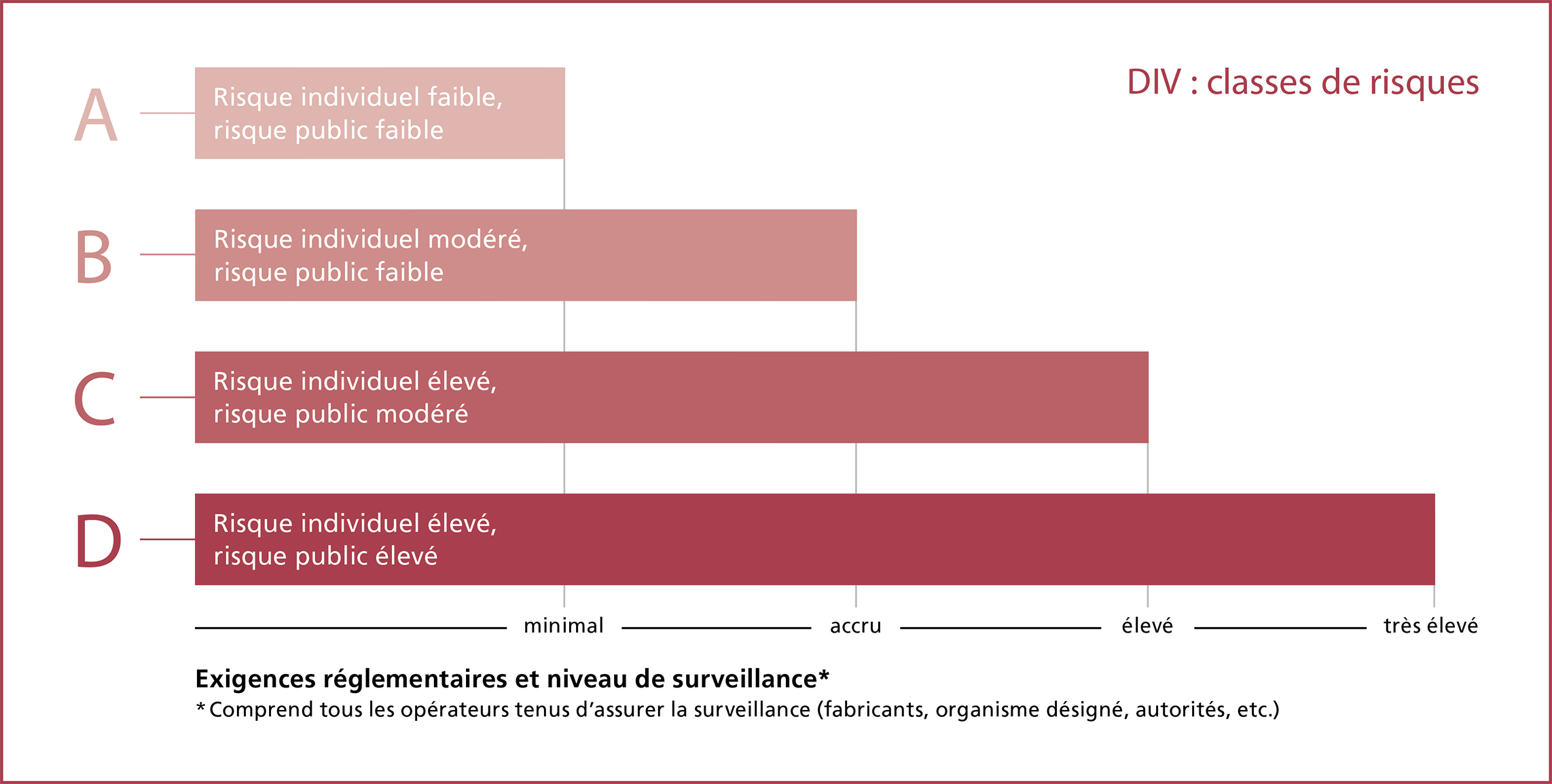
Les classes de risques
Avant toute mise sur le marché, un DIV est rattaché à l’une des quatre classes de risques définies (A, B, C et D), sachant que la classe A est celle des produits à faible risque (voir graphique). « Ce nouveau système de quatre classes implique notamment que les fabricants doivent recourir à un organisme désigné pour un nombre beaucoup plus élevé de DIV qu’avant, pour faire évaluer leur conformité », indique Einat Schmutz. Alors qu’auparavant, une grande partie des DIV pouvaient être commercialisés sur une simple déclaration du fabricant, ce n’est plus le cas aujourd’hui que pour les dispositifs de classe A, qui représentent environ 20 % de l’ensemble des DIV. Pour les dispositifs des classes B, C et D, il faut faire appel à un organisme désigné. Ce nouveau dispositif a principalement pour but d’assurer la sécurité des produits. « L’un des aspects clés du processus consiste à évaluer les performances cliniques. Il faut pour cela collecter des données cliniques dans le cadre d’études de performances. Ce qui compte, c’est la sécurité des patient(e)s », explique encore Einat Schmutz, exemple à l’appui : « Suite au durcissement des exigences, les tests génétiques et de dépistage du cancer sont passés de la catégorie la plus basse à la deuxième la plus élevée. »


