Beispiele Medizinprodukte
Was haben medizinische Implantate, Dialysegeräte, Herzschrittmacher, Dentalprodukte, Pflaster und Verbände, Sehhilfen, Röntgengeräte, Kondome, Schwangerschaftstests, Laborgeräte sowie Covid-19-Tests gemeinsam? Es sind Medizinprodukte, die von Swissmedic überwacht werden. Bevor sie in Verkehr gebracht werden, müssen sie bestimmte Anforderungen erfüllen und ein sogenanntes Konformitätsbewertungsverfahren durchlaufen. Bis zu einer halben Million verschiedene Produkte sind auf dem Markt erhältlich.
Beispiele In-vitro-Diagnostika
In-vitro-Diagnostika (IVD) sind eine Untergruppe der Medizinprodukte. «In-vitro-Diagnostikum bezeichnet ein Medizinprodukt zur In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben wie zum Beispiel Blut oder Urin. Dabei handelt es sich um Tests, die zum Beispiel auf biologischen Untersuchungen basieren und helfen, den Gesundheitszustand eines Menschen zu kontrollieren, etwa Brustkrebstests, Blutzuckertests oder auch HIV-Tests», erklärt Evelyn Aeschlimann. «Hersteller mit Sitz in der Schweiz müssen ihre IVD-Produkte melden, wenn sie die Produkte erstmals in der Schweiz in Verkehr bringen.»
Die Bedeutung
Die Wichtigkeit des Themas lässt sich daraus erkennen, dass der Bund eine spezifische und separate rechtliche Basis für IVD kreierte. «Diese Basis bilden zwei Bundesratsverordnungen mit rechtlichen Bestimmungen. Das war ein grosses Projekt, das mehrere Personen von verschiedenen Behörden während Monaten beschäftigte», erinnert sich Andreas Schlegel. Federführend in diesem Verfahren war das Bundesamt für Gesundheit (BAG). «Swissmedic wurde vor allem beigezogen, um die Umsetzbarkeit im Vollzug zu prüfen.»
Der Marktzugang
500 000 Medizinprodukte – davon rund 40 000 IVD. Im Vergleich: Im Bereich Humanarzneimittel existieren in der Schweiz rund 5700 zugelassene Arzneimittel. «Anders als bei Arzneimitteln gibt es für Medizinprodukte keine behördliche Zulassung, denn das würde den Rahmen sprengen, sondern das System der Konformitätsbewertung. Bei diesem Verfahren wird nachgewiesen, dass die geltenden Sicherheits- und Leistungsanforderungen erfüllt sind und ein akzeptables Nutzen-Risiko-Verhältnis besteht», erklärt Einat Schmutz. Bei Produkten höheren Risikos muss bei diesem Verfahren eine bezeichnete Stelle beigezogen werden. Die Swissmedic kommt bei der Bezeichnung und Überwachung dieser Stellen ins Spiel.
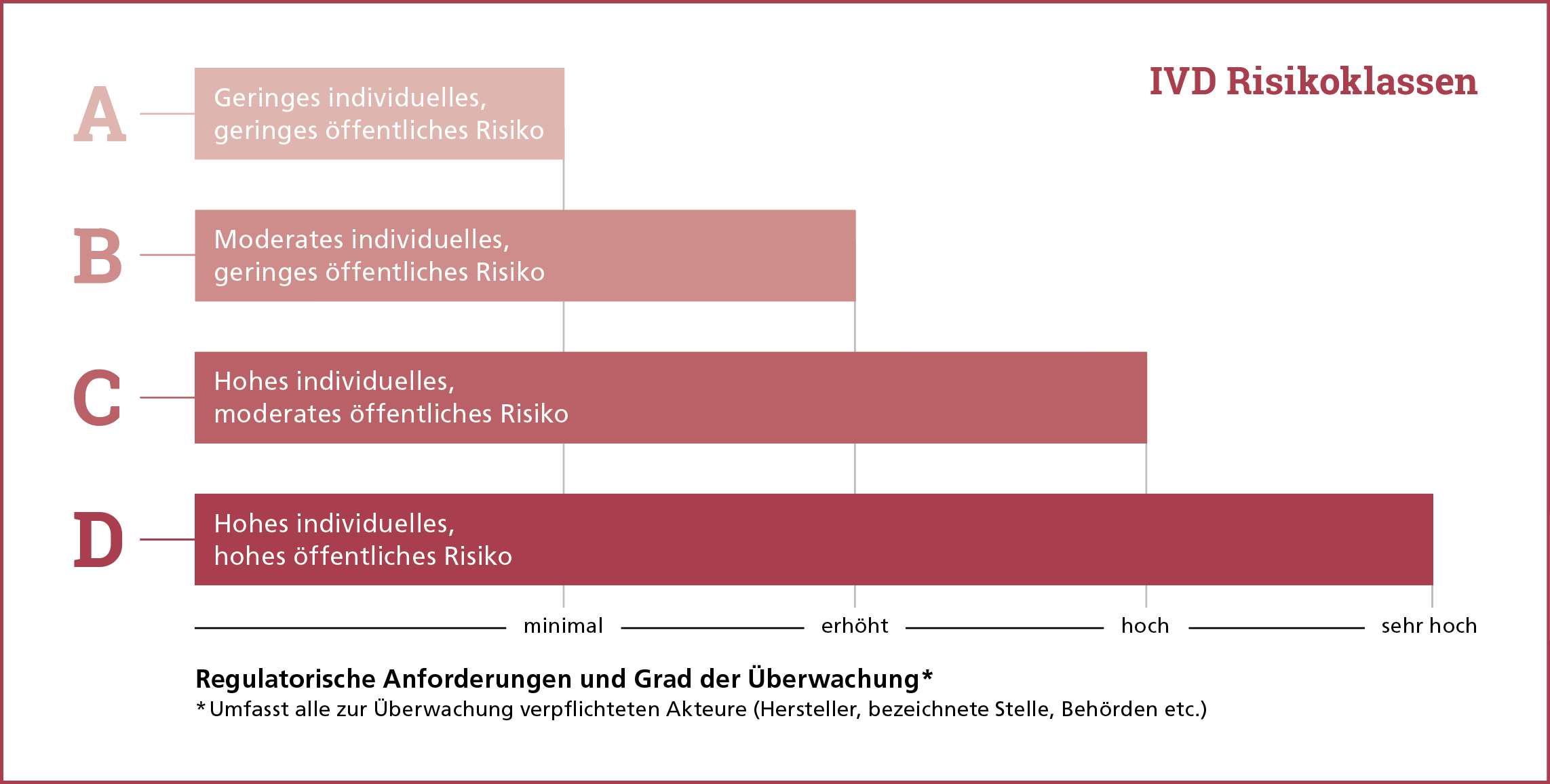
Die Risikoklassen
Jedes IVD wird vor dem Inverkehrbringen in eine von vier Risikoklassen (A, B, C, D) eingeteilt, wobei die Klasse A Produkte mit einem geringen Risiko umfasst (siehe Box). «Dieses neue Vierklassensystem bringt insbesondere mit sich, dass im Rahmen der Konformitätsbewertung für erheblich mehr IVD als bisher eine bezeichnete Stelle beigezogen werden muss», erklärt Schmutz. Wo früher ein Grossteil der IVD mit einer reinen Selbstdeklarierung des Herstellers auf den Markt gebracht werden konnte, gilt das heute nur noch für die Klasse-A-Produkte, die etwa 20 % aller IVD ausmachen. Für Produkte der Klassen B, C und D ist eine bezeichnete Stelle beizuziehen. Dabei geht es in erster Linie um die Sicherheit der Produkte. «Ein zentraler Aspekt ist insbesondere die klinische Leistungsbewertung. Dafür müssen klinische Daten im Rahmen von Leistungsstudien erhoben werden. Im Fokus steht die Patientinnen- und Patientensicherheit», erklärt Einat Schmutz und präsentiert ein Beispiel aus dem Alltag: «Aufgrund der strengeren Anforderungen haben die Gen- und Krebstests in den Risikoklassen einen Sprung von der untersten in die zweithöchste Kategorie gemacht.»


